Data Import into ggKbase
After the data preparation steps are completed, you will be left with a directory that looks similar to this (note that all files of interest have the common prefix BASE_NAME_scaffold_min1000* ):
> ls begin bt2 BASE_NAME_scaffold.fa BASE_NAME_scaffold_min1000.16S.cmsearch BASE_NAME_scaffold_min1000.fa BASE_NAME_scaffold_min1000.fa.16s BASE_NAME_scaffold_min1000.fa.genes BASE_NAME_scaffold_min1000.fa.genes.faa BASE_NAME_scaffold_min1000.fa.genes.faa-vs-kegg.b6+ BASE_NAME_scaffold_min1000.fa.genes.faa-vs-kegg.b6.gz BASE_NAME_scaffold_min1000.fa.genes.faa-vs-uni.b6+ BASE_NAME_scaffold_min1000.fa.genes.faa-vs-uni.b6.gz BASE_NAME_scaffold_min1000.fa.genes.faa-vs-uniprot.b6+ BASE_NAME_scaffold_min1000.fa.genes.faa-vs-uniprot.b6.gz BASE_NAME_scaffold_min1000.fa.genes.fna BASE_NAME_scaffold_min1000.fa.trnascan BASE_NAME_scaffold_min1000.fa.trnascan.fasta contig.fa end log mapped.log
>> Ask Rohan to move the final import files to assembly.d in the project’s directory within /group/banfield/sequences/2020/. This is where they should be imported from.
All the BASE_NAME_scaffold_min1000* files, with their properly formatted names, are used during the ggKbase import steps…
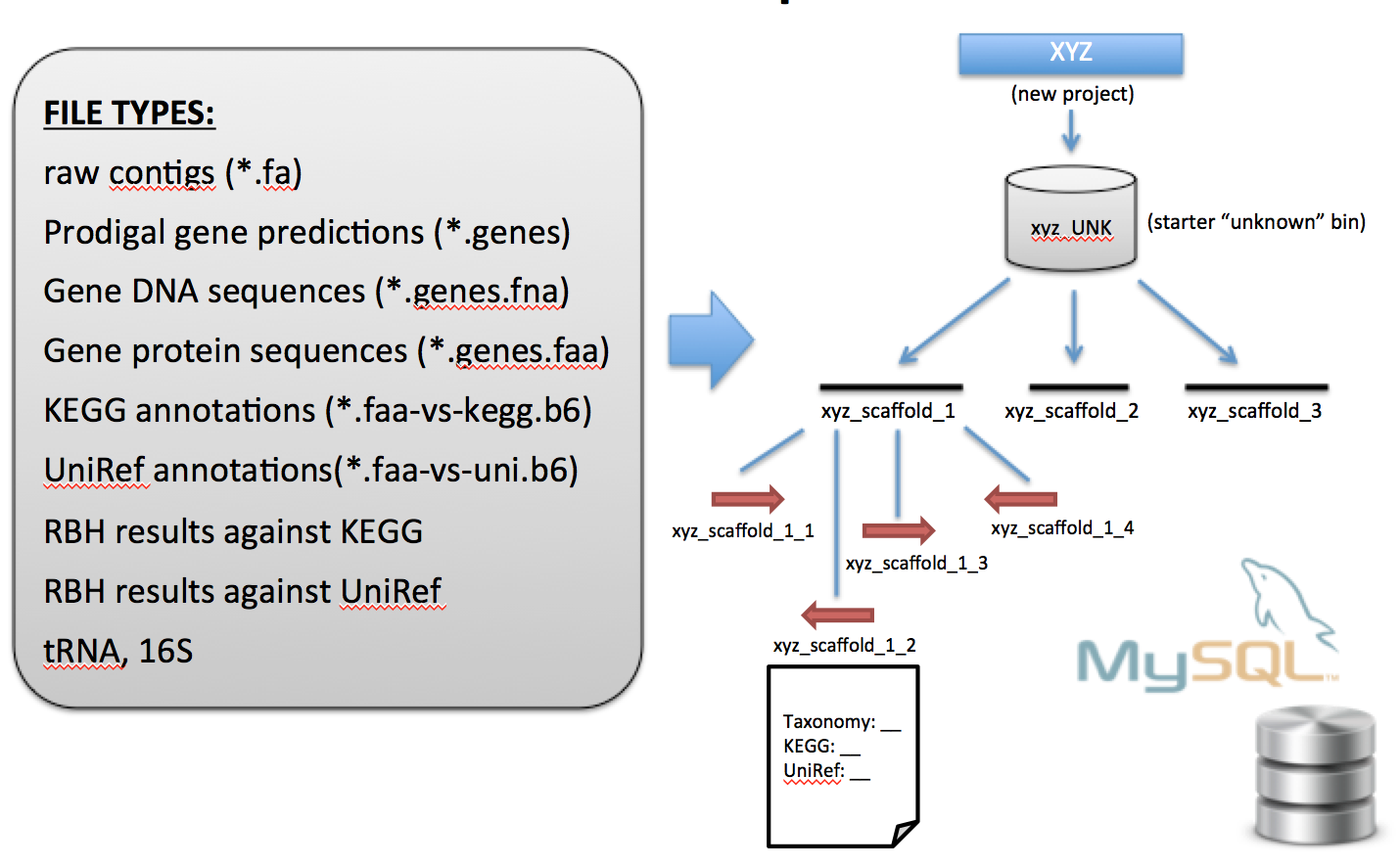
Schematic diagram for importing data files into the ggKbase database.
Importance of Sample Metadata
We are trying to pay more attention to metadata associated with each sample. Instead of keeping these details in a spreadsheet, away from the data in ggKbase, we’re incorporating it into the system. This is an ongoing discussion – if you think you have some metadata that belongs in ggKbase, submit a ticket.
Project Creation
If you have a small number of projects to create and are a member of the Banfield research network, you can create new projects by going here: http://ggkbase.berkeley.edu/projects/new.
Follow the instructions there for filling in the metadata.
If you have many (e.g. >3) new projects to create:
To import your data, download the template below and complete it with your sample information. Save the file as a TSV (in Numbers, go to File > Export To > TSV) and send the completed table to Leylen via Slack.
ggkbase_bulk_upload_template_2025
**NOTE: Exporting as a .tsv from excel messes up the formatting, please use “Numbers” in Mac ideally or Google sheets to fill in your info.
To find total basepairs assembled:
On biotite, use
sum-bp *min1000.fa
To find total read basepairs:
- If you used IDBA for assembly: Find the number of reads in the second line of the idba_ud ‘log’ file. Multiply this number by the read length.
- If you used another assembler use:
sum-bp /groups/banfield/sequences/2025/<BASE_NAME>/raw.d/*_trim_clean.PE.*.fastq.gz
Instructions for TSV Validation Before Sending
1. Run the following command to inspect the TSV:
python /home/leylenm/scripts/ggkInspector.py -t path/to/tsvReplace path/to/tsv with the actual file path.
Note: If you don’t have them, install the necessary packages to be able to run the script:
pip3 install mysql mysql-connector-python pandas2. If the script detects errors, fix them before sending the file.
•Make all necessary corrections and rerun the script until there are no errors before sending it to Leylen.