Guide to NCBI WGS Submissions
Intro:
Join Banfield Lab NCBI. Slack Lily your email to be added to the group.
Our lab’s contact is Larissa Brown. You may not email her directly until she contacts you, then you can reply to genomes@ncbi.nlm.nih.gov.
Have NCBI annotate your genomes. Do not attempt to submit annotations unless absolutely necessary.
ALWAYS use Banfield Lab affiliation to submit (not your personal account.)
NCBI’s resource page:
https://www.ncbi.nlm.nih.gov/genbank/wgsfaq/#metagen
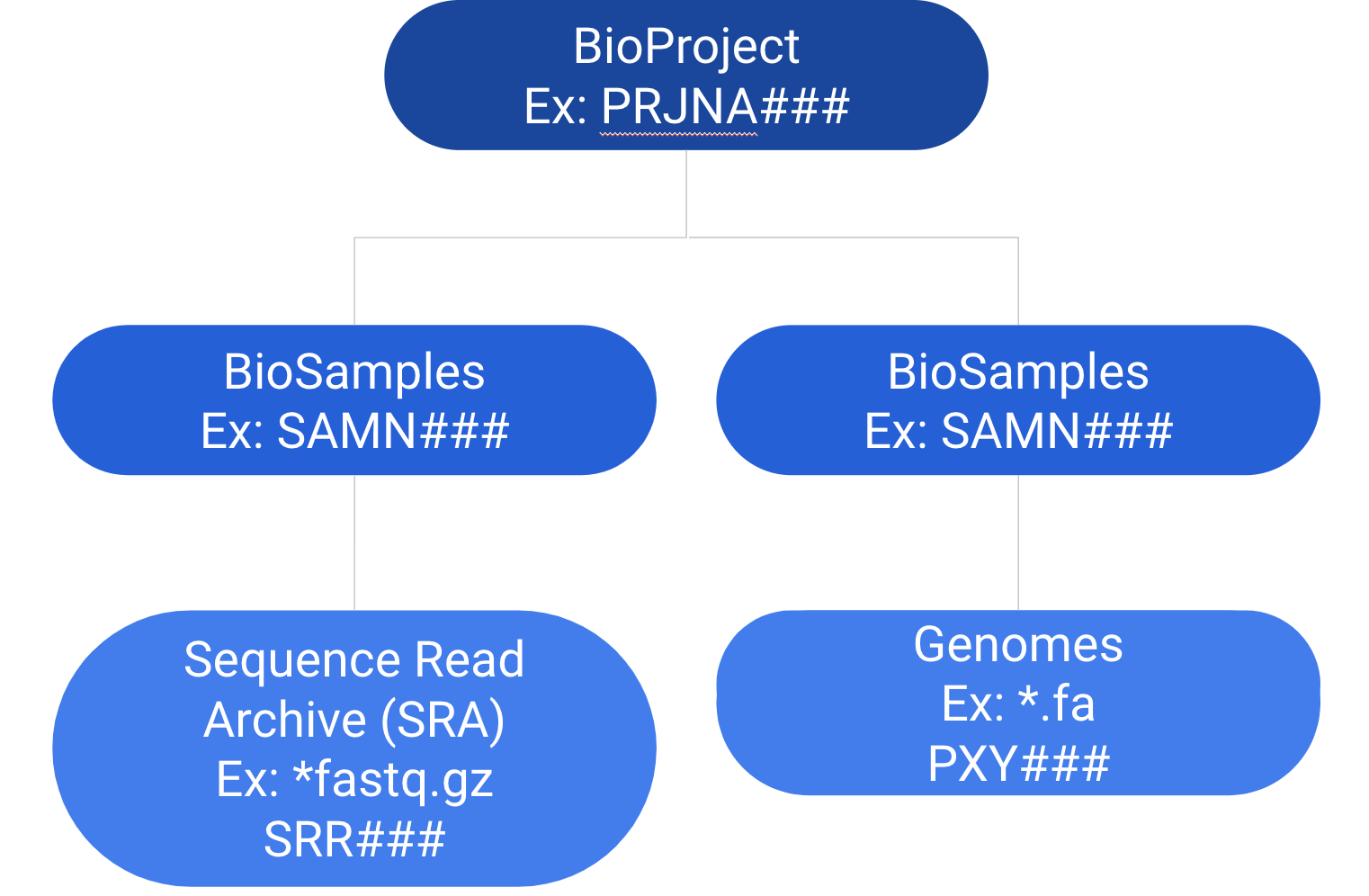
Hierarchical Flow of NCBI:
Steps to Submit:
Submission portal link: https://submit.ncbi.nlm.nih.gov/subs/
Note: When linking to a publication, do not use the doi or some number to link it, this has always pulled a completely random publication from the NCBI database. I advise to fill in the title, authors, etc manually.
Create BioProject
Skip this step if there is already a BioProject. The BioProject Number is PRJNA#### and may be found in the related publication.
From the Submission Portal (link above), select BioProject beneath “Start a New Submission”
This should be pretty straightforward. Let me know if you have any questions, thoughts on what should be added to this help page.
Usually we select for project type: genome sequencing and assembly and for sample scope: environment.
Create Biosamples
From the Submission Portal (link above), select BioSample beneath “Start a New Submission”
You will have to make TWO different kinds of Biosamples for both SRA and for genomes.
MIMS and other “metagenome” samples are expected to have a “metagemone” organism name. Assemblies from a metagenome, on the othe hand, will have the bacterial (or other) organism name and should be submitted with the MIGS.ba package.
FOR SRA: Follow the steps. Select “MIMS” for metagenome/SRA (use the 4.0 package.) https://www.ncbi.nlm.nih.gov/biosample/docs/packages/

For “organism” write “Environmental Metagenome,” for “host” write “Earth’s crust,” for “isolation_source” write “biofilm” (or what is appropriate.)
FOR GENOME: Use MIGS.ba package. For “organism” write species name. Any organism name that ends in “sp.” is automatically flagged for curator review. For cultured organisms, NCBI requires a species name, but not for genomes assembled from metagenomes, so those names are correct as they are. They just require manual review. You can ignore the warnings and click “Continue” again to move on with the submission.
NCBI will process your biosamples, and once you have the SAMN numbers you can move on to the uploads.
Just put “missing” or “not applicable” for spreadsheet columns which are not relevant.
Upload Raw Reads:
From the Submission Portal (link above) Select Sequence Read Archive beneath “Start a New Submission”
Fill in the spreadsheet for batch upload.
In regards to uploading your data:
We recommend using ftp in the background from a tmux. Select “Preload folder.” Follow instructions and create subdirectory in upload directory on the NCBI server. (It will not recognize files in your ncbi-home-upload directory.)
If you have many files, and you don’t want to tar them together, sometimes the ftp can fail at iterating through files. Try using mput instead of put, and also `mput` asks for a prompt each time, `y or n` to transfer a file. If you type `ftp -i <server name>` it will turn interactive mode off, and never ask for any prompts.
Upload Genomes:
First you must format your fasta files. All of the headers need to contain:
[organism=X] [isolate=X]
For example:
>qh_10_scaffold_989 id=10405260 bin="QH_10_Halobacteriales_67_13" [organism=Halobacteriales archaeon QH_10_67_13] [isolate=Halobacteriales archaeon QH_10_67_13]
This organism and isolate information matches your spreadsheet. This should be a quick script to edit these fas, but if you’d like to look at something I hacked together ask me (Kate.)
Next create a tarball of your genomes:
tar -cvzf fa_files.tgz fa_files/
Follow same upload instructions as above in “Upload Raw Reads” section.
If NCBI emails you back with a file containing contamination info about your genomes, Patrick West wrote a script to remove the contamination. See here:
https://github.com/patrickwest/NCBI_contamination_fix
Note: In the past people in the lab have had to “Prescreen” and have used sqn files instead of fa files. I highly recommend avoiding this and have had no problem with tarballs of simple *.fas.
However these are some notes from previous lab members on how to do this:
Part 1 – Prescreen.
Create a tarball of genomes (fasta is ok) and submit it to the WGS pipeline. NCBI will run a prescreen and identify contamination, adaptors, and duplicate seqs. Remove all contamination and adaptors sequences (duplicates are up to you).
Part 2 – Submission files (1 SQN file for each genome)
Create a template for submission (Authors, Paper title etc.) https://submit.ncbi.nlm.nih.gov/genbank/template/submission/
Create a structured comment template (Assembler, Coverage): https://submit.ncbi.nlm.nih.gov/structcomment/genomes/
Download tbl2asn and sequin from the NCBI ftp site.
Add organism names to contig headers like this:
>RBG_13_scaffold_20822_curated [organism=RBG_13_WWE3_37_7_curated]
Run tbl2asn as follows:
tbl2asn -p (directory of fasta files)-r (Output directory)-a s –t template.sbt -w structured_comment.asm
For tbl2asn – Fasta file should have a .fsa suffix only!
Create spreadsheet (similar to as described above).
Create tarball of sqn files and upload on WGS Submission page. Send NCBI Genomes your spreadsheet to link genomes with files.