A post by Lin-Xing Chen, Karthik Anantharaman, Alon Shaiber, A. Murat Eren, and Jillian F. Banfield. For any questions about the methods described below, please contact us via email (linxingchen@berkeley.edu).
Introduction
The purpose of this post is to describe step-by-step procedure for scaffold extension and gap closing to dramatically improve the quality of metagenome-assembled genomes (MAGs). These strategies are described in our recent paper entitled “Accurate and Complete Genomes from Metagenomes” (“CGM” for short hereafter), which is available at https://genome.cshlp.org/content/early/2020/03/18/gr.258640.119.full.pdf+html. Please cite this publication if you follow the procedures described here for your analyses.
The CGM paper summarizes the history of genome-resolved metagenomics, with examples from publications, to show the necessity of genome binning and manual curation of MAGs to avoid misleading conclusions. Our paper provides a step-by-step procedure of how to curate a draft MAG generated by manual or automatic binning tools to prevent errors (e.g., misbinned scaffolds, local assembly errors), close scaffolding gaps and thus generate high-quality or complete MAGs (CMAGs). Here we will detail the two primary strategies we commonly use to improve MAGs to completion: scaffold extension and gap closing.
Scaffold extension
Scaffold extension uses the paired-end reads mapped to a given scaffold to extend its ends, (1) to get the complete sequence of a protein-coding gene or rRNA gene at the ends, (2) to assemble it with another scaffold based on overlap sequences at the end, (3) to obtain a complete genome of bacteria, archaea, virus, phage, etc. The CGM paper says,
“Scaffolds within a bin that do not overlap at the start of curation may be joined after one or more rounds of scaffold extension. This process of extending, joining and remapping may continue until all fragments comprise a single circularized sequence. It should be noted that read by read scaffold extension is very time-consuming. If an extended scaffold cannot be joined to another scaffold after a few rounds of extension it may be worth testing for an additional scaffold (possibly small, thus easily missed by binning) by searching the full metagenome for overlaps. Sometimes, the failure of scaffold extension is due to missing paired reads, which may be found at the end of another fragment. If they are pointing out but the sequences cannot be joined based on end overlap, a scaffolding gap can be inserted in the joined sequence (reverse complementing one of the scaffolds may be necessary).”
This means that sometimes it is possible to link all the scaffolds of a draft MAG into a circular genome by scaffold extension followed by overlap-based assembly of the extended scaffolds. However, it should be noted that a manual scaffold extension is time-consuming and often does not lead to a circular MAG genome sequence. Additional tests should be performed to verify the accuracy of the final product as we described in our study.
How can scaffold extension be performed?
We manually extend the scaffolds in a given bin to be curated using Geneious. There are several steps for this, including (1) mapping of reads to the scaffolds that comprise a bin, (2) visualization in Geneious and (3) manual extension in Geneious. Note that the reads used for extension should be appropriately placed relative to their already mapped pair, in the right orientation, and the originally mapped pair should support the consensus sequence at the scaffold ends. The following sections will detail each of these three steps.
(1) Reads mapping
Read mapping can be performed with available tools, for example, Bowtie 2. Here, we illustrate using the first genome bin (comprising a single scaffold) described in the section of “Case studies illustrating the curation of draft MAGs” in CGM, i.e., ALT_04162018_0_2um_scaffold_13. The scaffold was assembled from paired-end reads (ALT_04162018_0_2um.1.fastq.gz and ALT_04162018_0_2um.2.fastq.gz) and is in FASTA format. Two steps are needed for read mapping using Bowtie 2.
First, build the database for mapping.
bowtie2-build ALT_04162018_0_2um_scaffold_13.fasta ALT_04162018_0_2um_scaffold_13.fasta
Second, map the reads to the scaffold
bowtie2 -p 6 -X 2000 -x ALT_04162018_0_2um_scaffold_13.fasta -1 ALT_04162018_0_2um.1.fastq.gz -2 ALT_04162018_0_2um.2.fastq.gz | shrinksam -v > ALT_04162018_0_2um_scaffold_13.fasta.mapped.sam
Note: “-p 6” is the number of cores used for mapping, “-X 2000” is the largest insert length allowed when mapping paired-end reads, which could be modified based on the study. Shrinksam is a tool to filter the original SAM file from Bowtie 2, which will only write the mapped reads to the file and thus save a lot of storage space. Shrinksam could be downloaded from Github (https://github.com/bcthomas/shrinksam).
(2) Visualization in Geneious
Prepare the scaffold file (.fasta) and the Bowtie 2 mapping file (.sam), using the function in Geneious, “File” -> “Import” -> “From File”.
Once the fasta file is imported, select it and import the SAM file.

Select the appropriate .fasta sequence used for mapping:
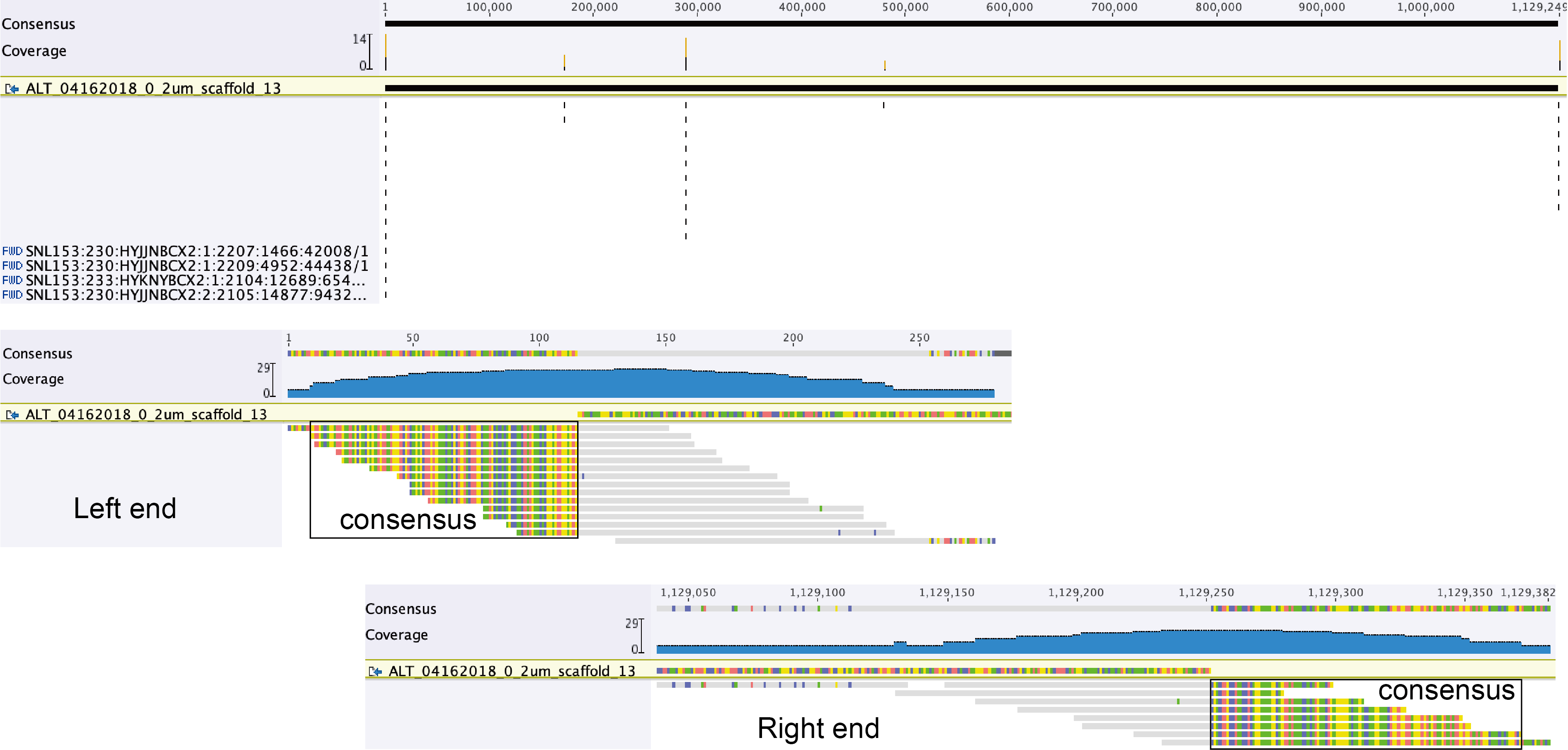
Two files are generated, one (the middle one in the screenshot below) shows the reads that are mapped to the scaffold, the other (the bottom one) includes the unplaced reads from pairs (means the other reads in those pairs were mapped to the scaffold via Bowtie 2).

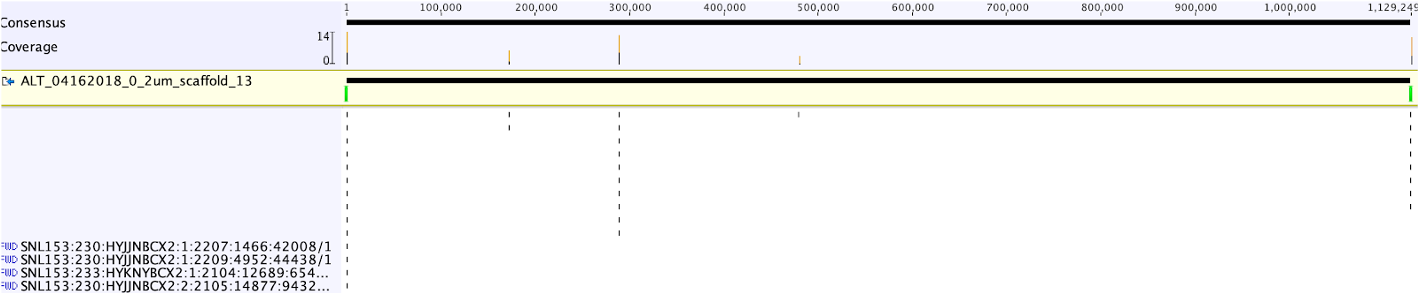
Below is the overview of all reads mapped to the scaffold, the coverage profile is generally even (except a region between 700,000 and 800,000 bp, which is a prophage region).

(3) Manual extension of scaffold
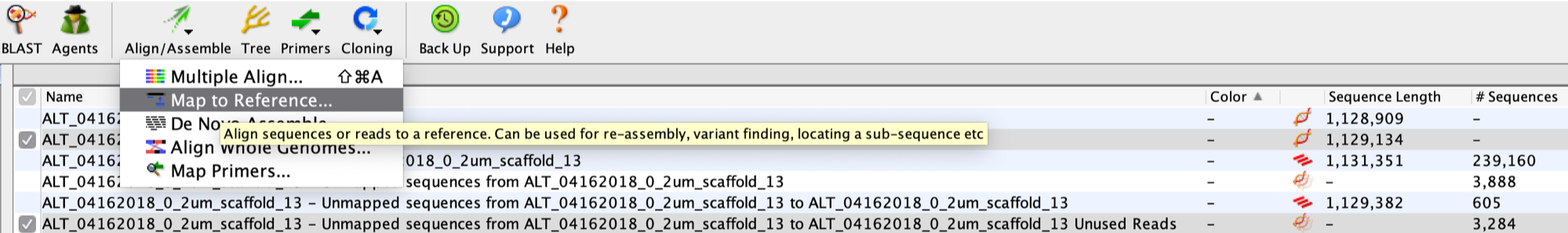
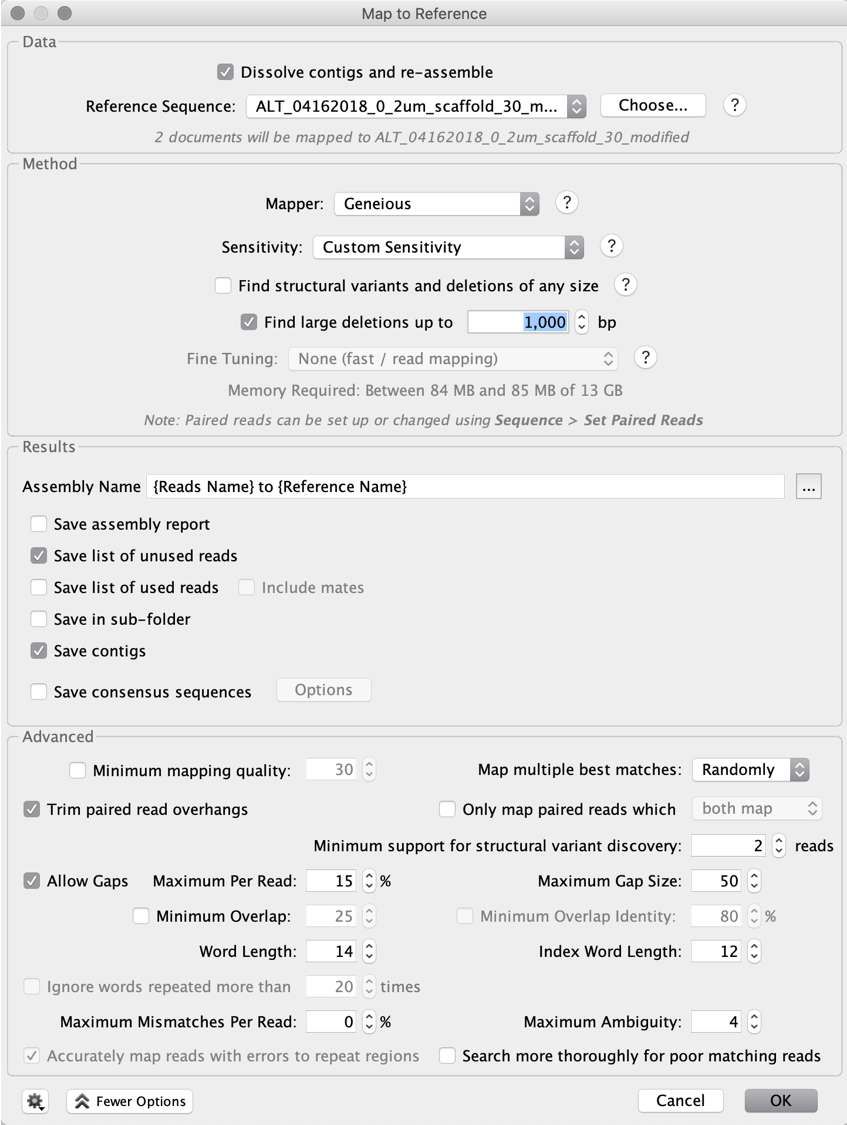
The next step is to extend the scaffold manually in Geneious. First, select the FASTA file and the unplace reads file (mentioned above), and use the Geneious function “Align/Assemble” -> “Map to Reference”.
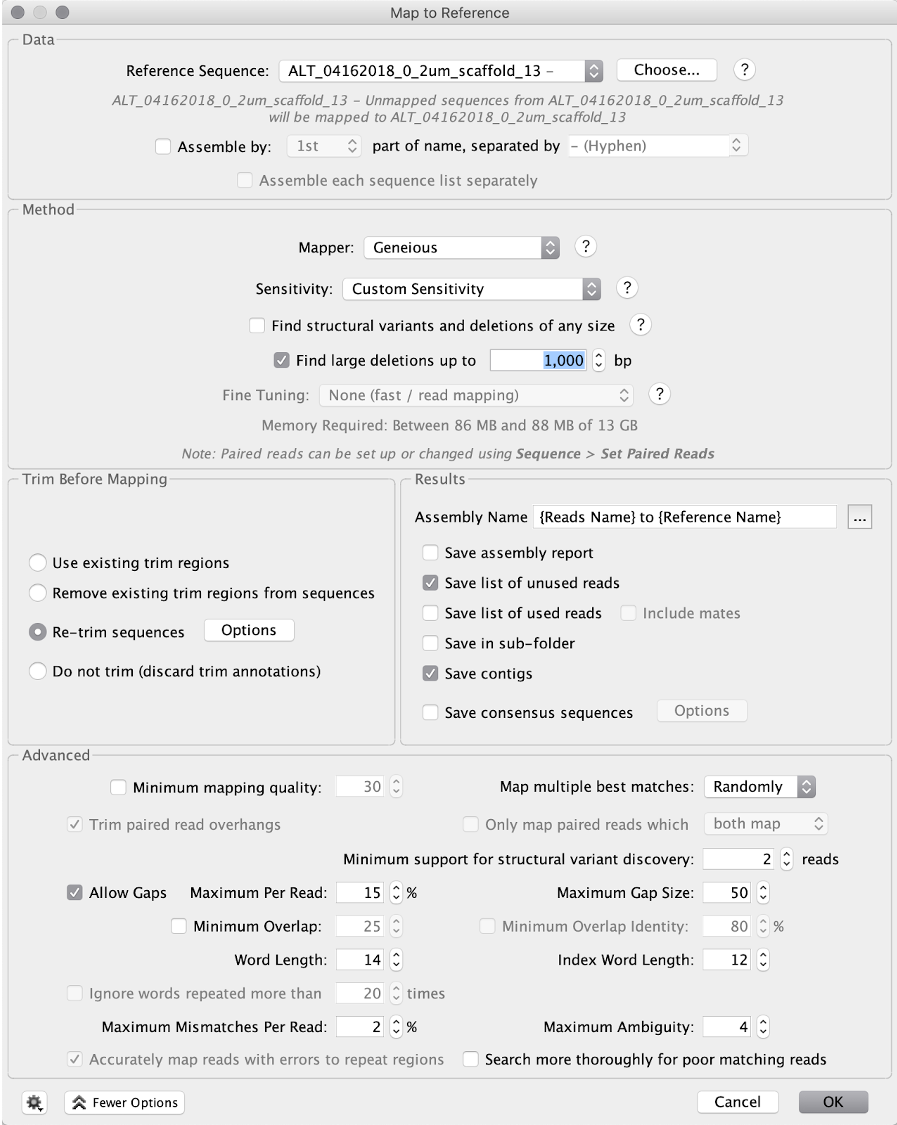
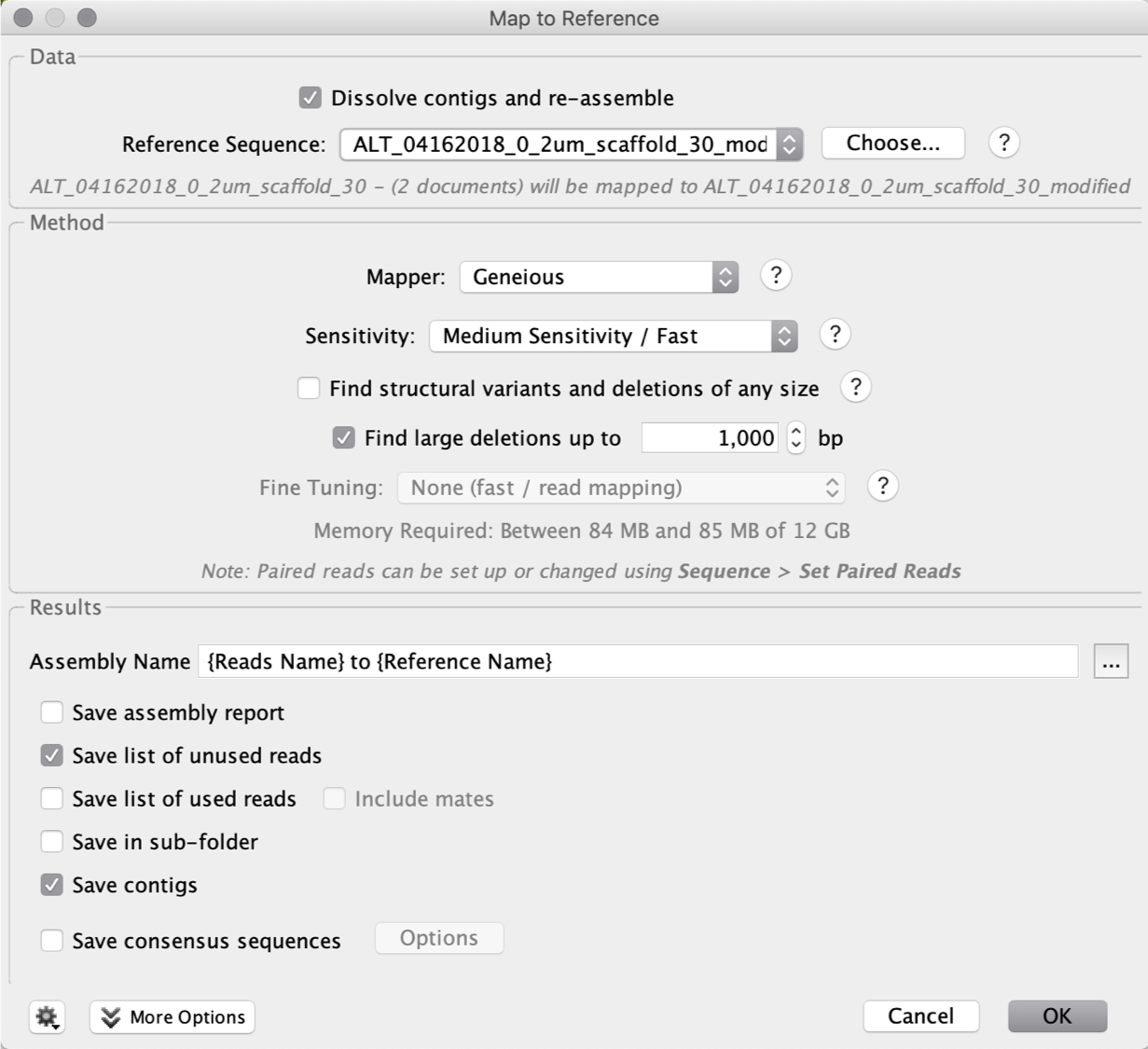
For sensitivity, use “Custom Sensitivity” and set “Maximum Mismatches Per Read” as 2%, do not change any other parameters. You can check “Save list of unused reads” so that the saved reads will be used for the next run of extension.
Two files are generated by this step (at the bottom of the screenshot below): one that contains the 605 unplaced reads Bowtie 2 mapped to the scaffold, and another one that contains the 3,284 reads that remain unmapped.

Check the reads mapped to the ends of the scaffold. The consensus sequences of reads that only partially mapped to the scaffold should be used for extension (see Left and Right ends, below: copy and paste the read sequence into the consensus sequence line above them).
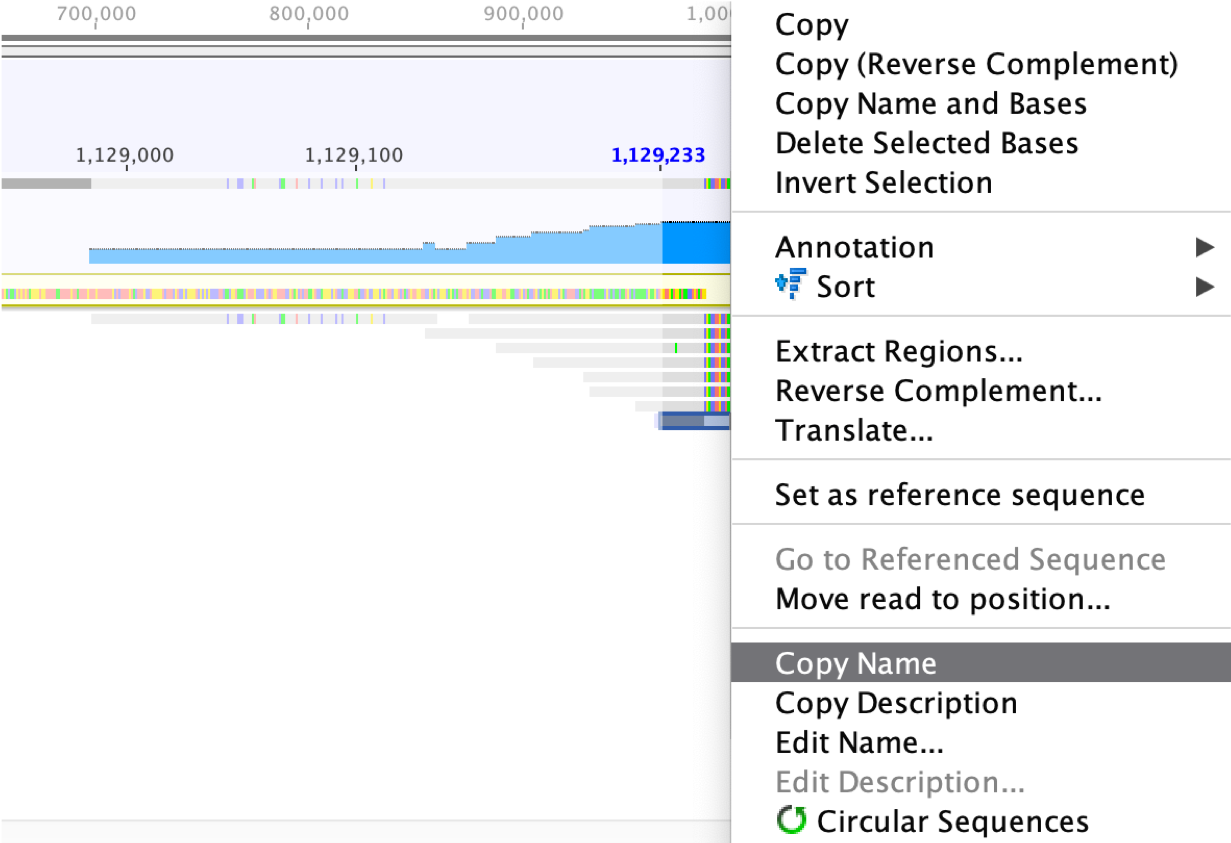
Before extending the scaffold, the reads in the pairs that already mapped to the scaffold should be checked to see if they are placed at an appropriate distance away, given the library size information. This can be done by searching the names of the reads. For example, the name of the most bottom read on the right end is “SNL153:233:HYKNYBCX2:1:1109:2879:14634/1 (reversed)”, which could be copied as shown in the screenshot below.
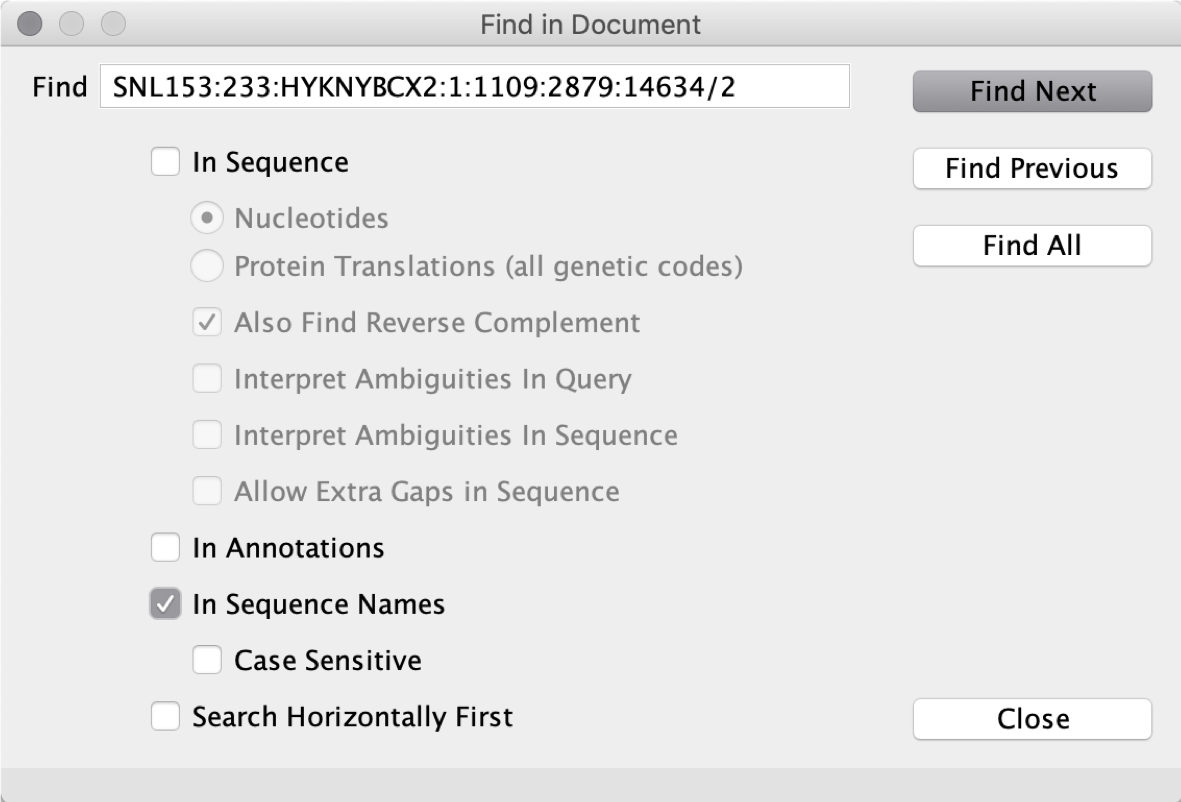
Thus, we should search “SNL153:233:HYKNYBCX2:1:1109:2879:14634/2” in the original SAM file from Bowtie 2 to check its mapping location and orientation (see below).
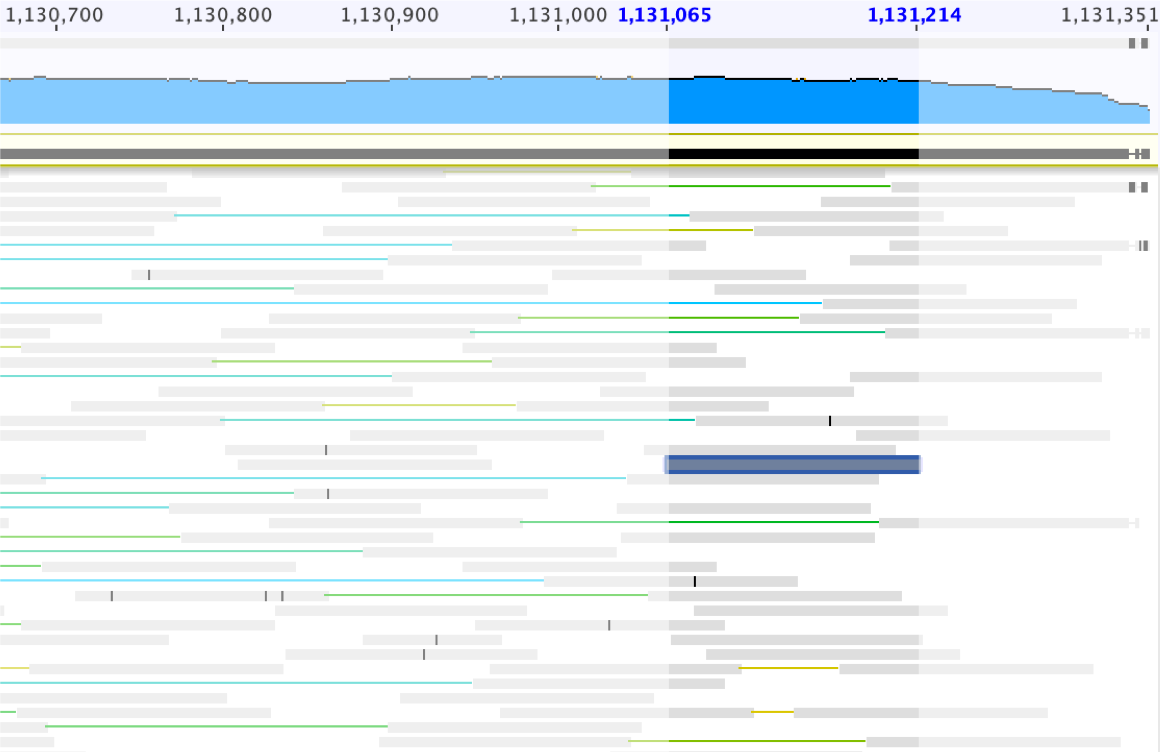
The extended scaffold sequence can be saved as “ALT_04162018_0_2um_scaffold_13_extended” (from 1,128,909 bp to 1,129,134 bp in length).

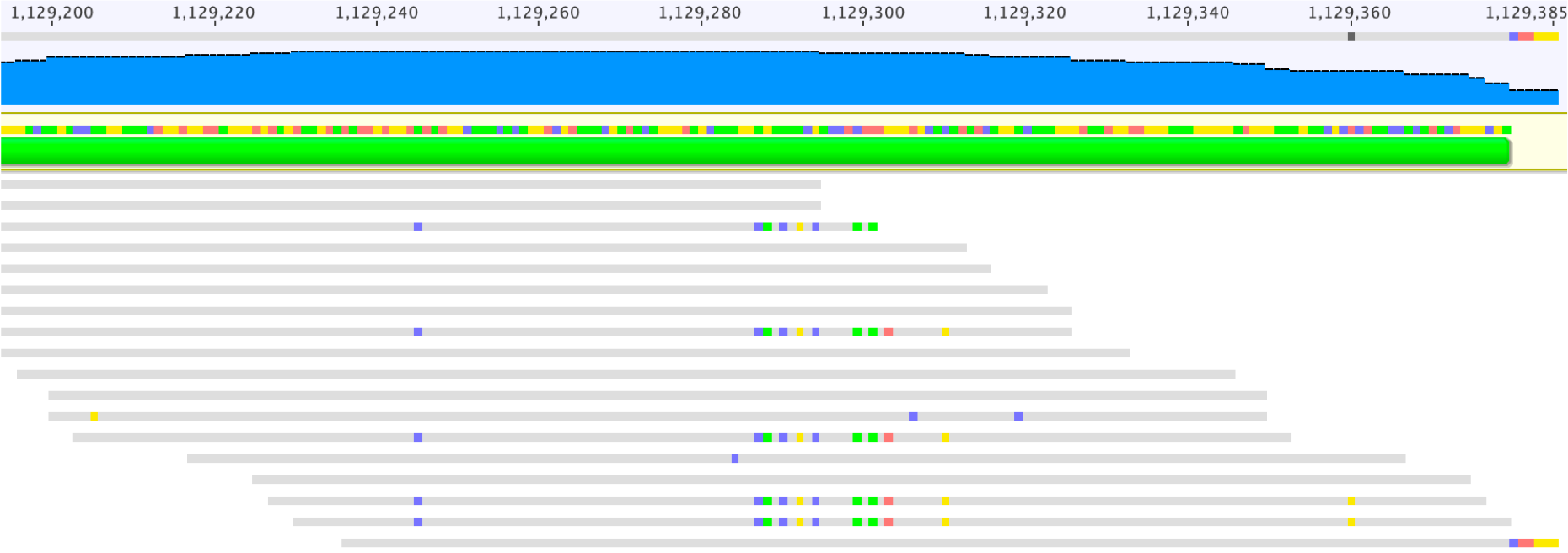
Then, map the unused reads set from the last mapping run to the extended scaffold. This should be repeated until no more unplaced reads are recruited. If necessary, the full metagenome read dataset can be remapped to the extended scaffold sequence to continue the process.

You may see variations in reads that map to scaffold ends which may indicate distinct paths or sequence variants (possibly due to the presence of subpopulations in the environment). As shown above, it may be possible to determine the correct path if only one of two variants has a paired read that supports the consensus. If this isn’t the case, you may choose to follow the variant path with the most appropriate coverage (or the majority of reads), or terminate the extension effort.
After a round of scaffold extension, it may be possible to find scaffolds in the genome bin that can be joined (in some cases, spanned by paired reads). If no scaffold is found, the sequence of the new scaffold ends can be searched against the entire metagenomic scaffold set to find a fragment that was not included in the bin (with the right GC content, coverage and phylogenetic profile). This sequence can be used to extend the scaffold and reads remapped for further validation.
Gap closing
When scaffolding is a step of de novo assembly (assemblers like IDBA_UD, metaSPAdes) of a given metagenomic dataset, Ns are inserted between contigs spanned by paired-end reads. CMAGs represent genomes without Ns, thus the N gaps in the scaffolds should be filled by the appropriate sequence.
How can gap closing be performed?
The first step should be to check whether the N gap is indeed spanned by paired reads (if not, the assembly may be chimeric). The second step should be to see whether the sequence in the gap region is already present but duplicated on either side of the Ns (thus, the gap can be closed by removing the duplicated region and the Ns).
Gap closing can be performed by automatic tools like Gapfiller (Nadalin et al. 2012), even though we have not evaluated its performance. Here, we describe an alternative strategy and show how this can be done using Geneious.
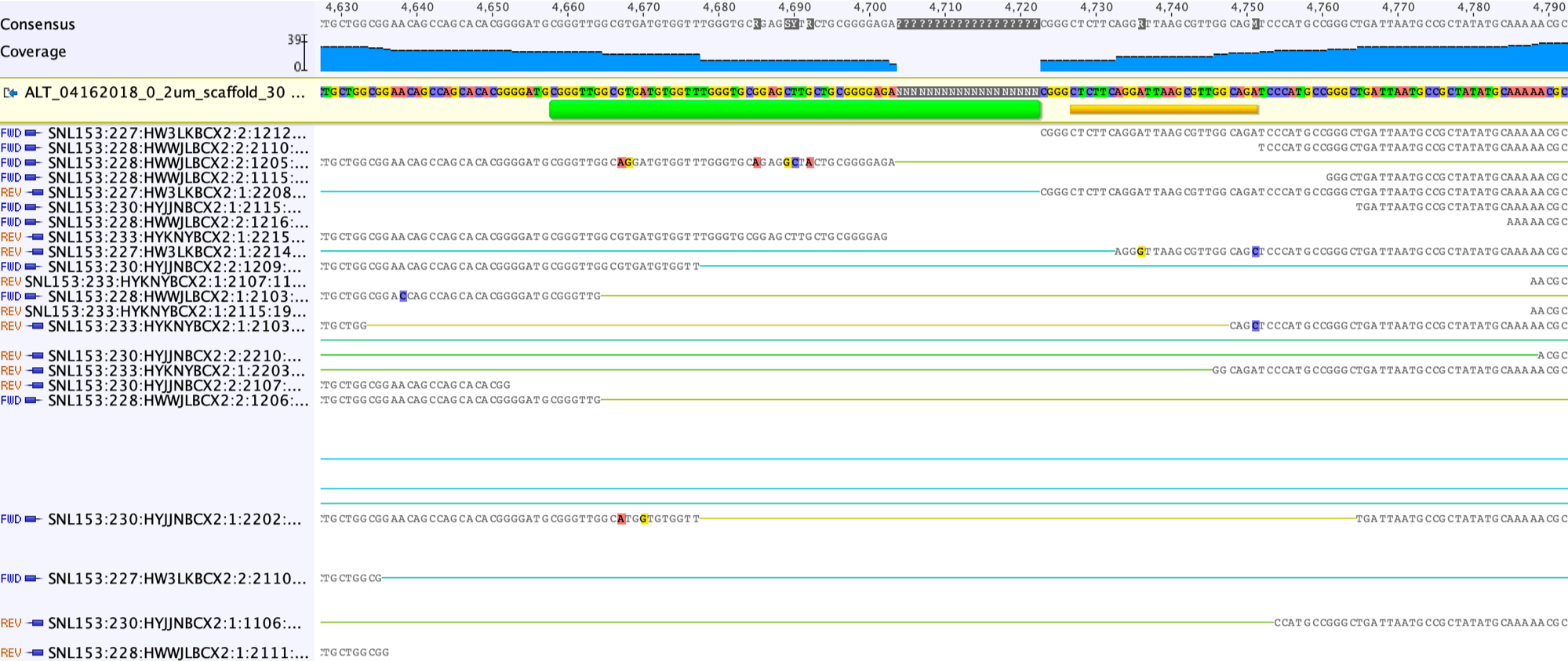
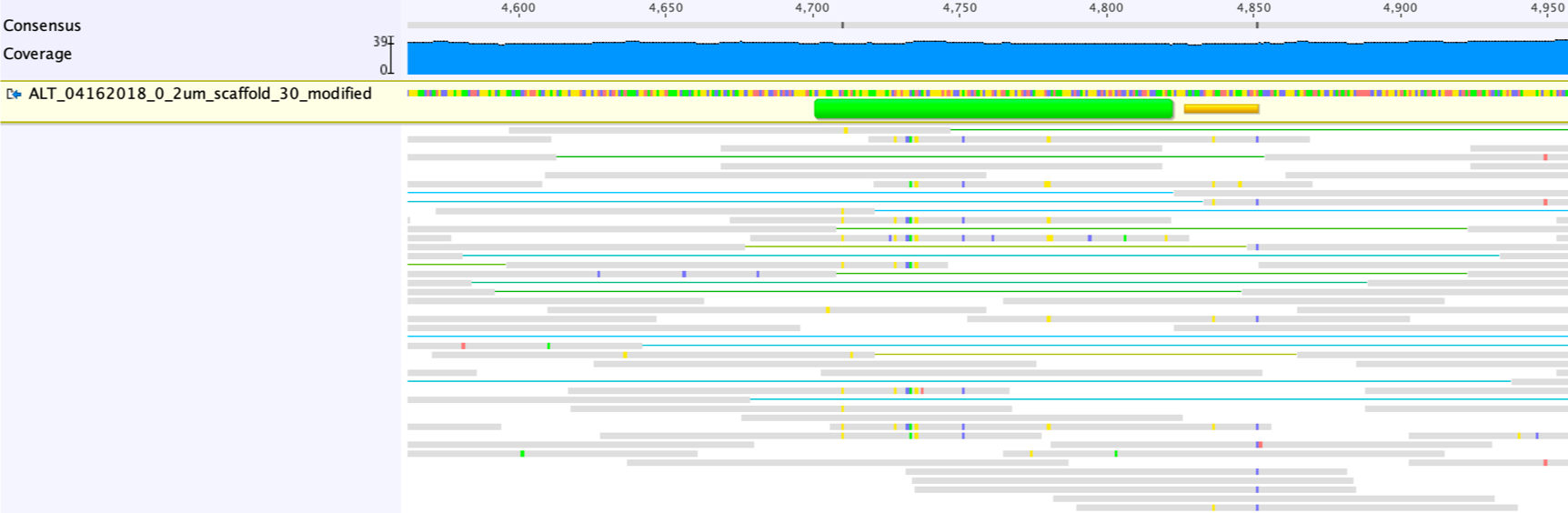
To illustrate, we use another scaffold from the same sample as the one we described above, i.e., ALT_04162018_0_2um_scaffold_30, which contains a scaffolding gap (these gaps can be quickly located by searching “Ns”).

The figure below demonstrates the read mapping profiles from Bowtie 2.

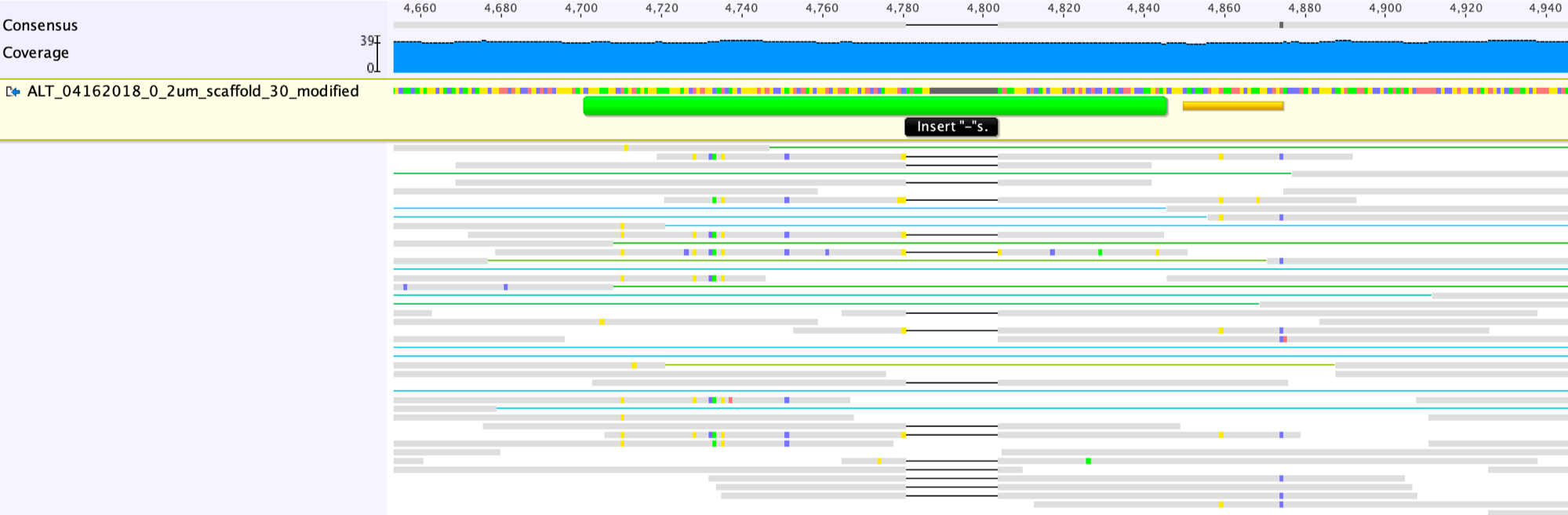
The first step to fix the scaffolding gap is to open the gap, by adding a suitable number of “-” into the Consensus sequence (the grey line in the screenshot below shows the right place to add the “-“).
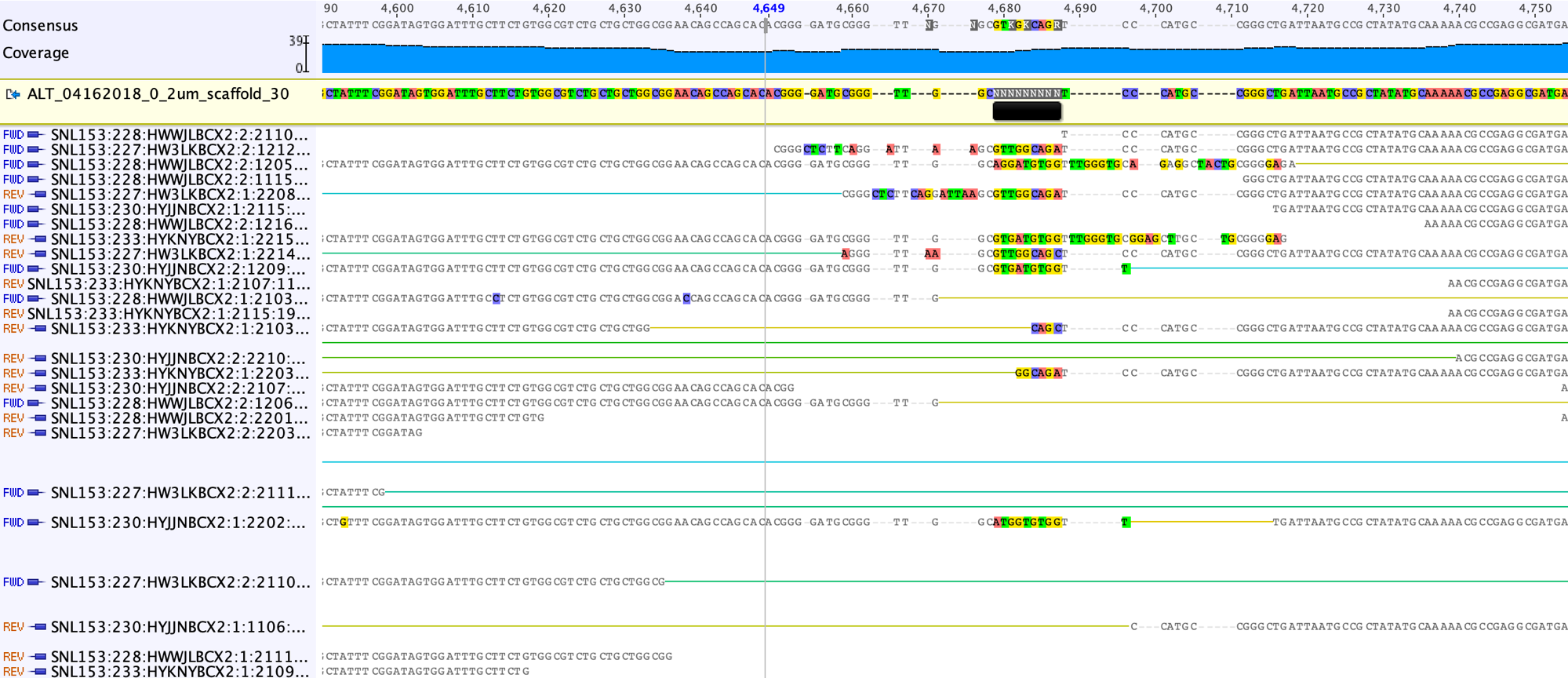
This is what looks like after adding the “-” to the left of the region of confusion.
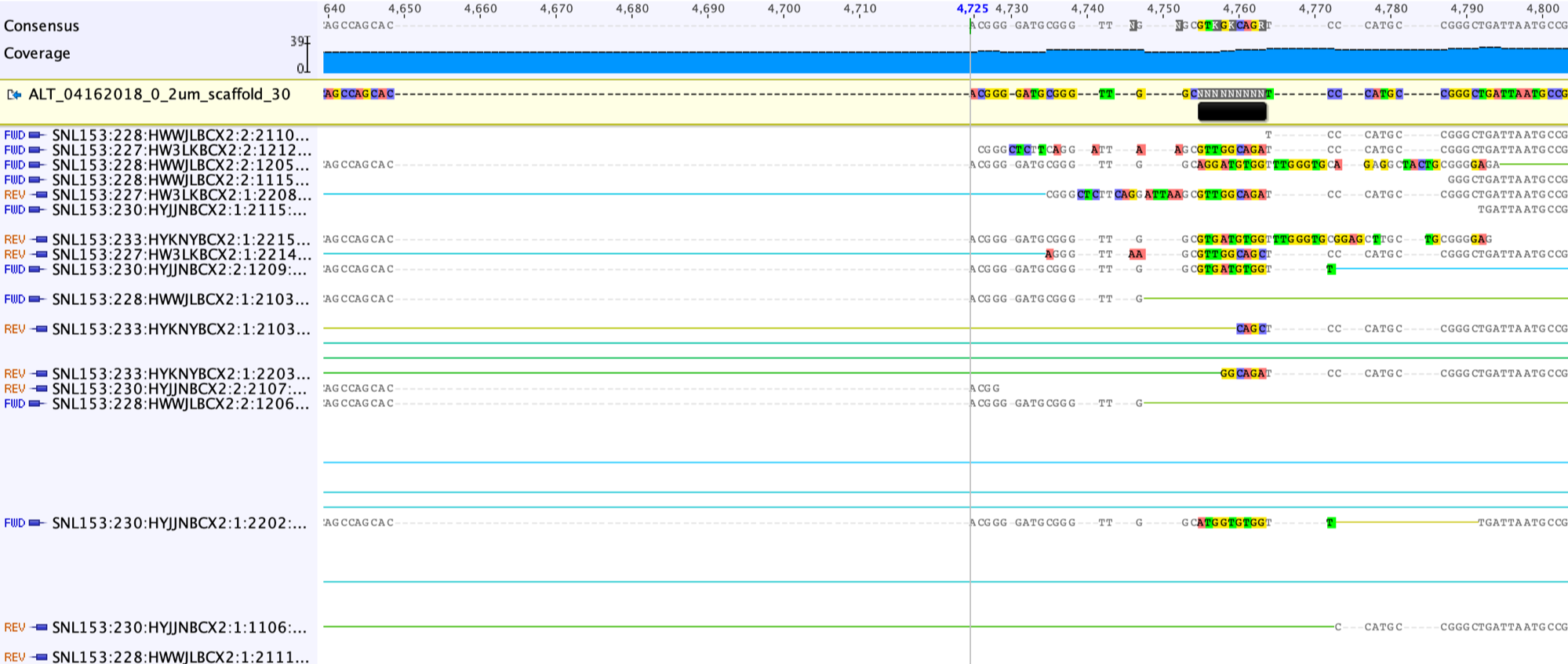
Then we should move reads in this region to the appropriate side:
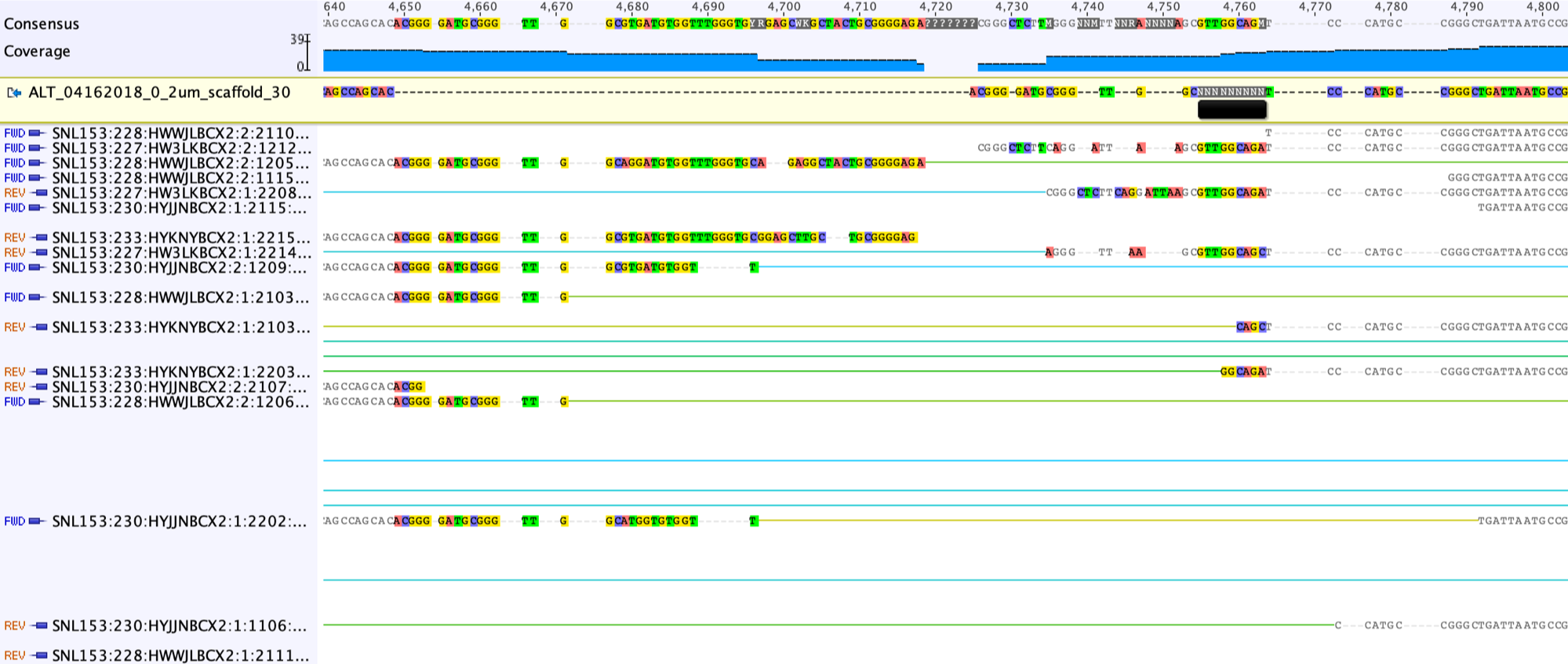
Then delete the columns without any base.
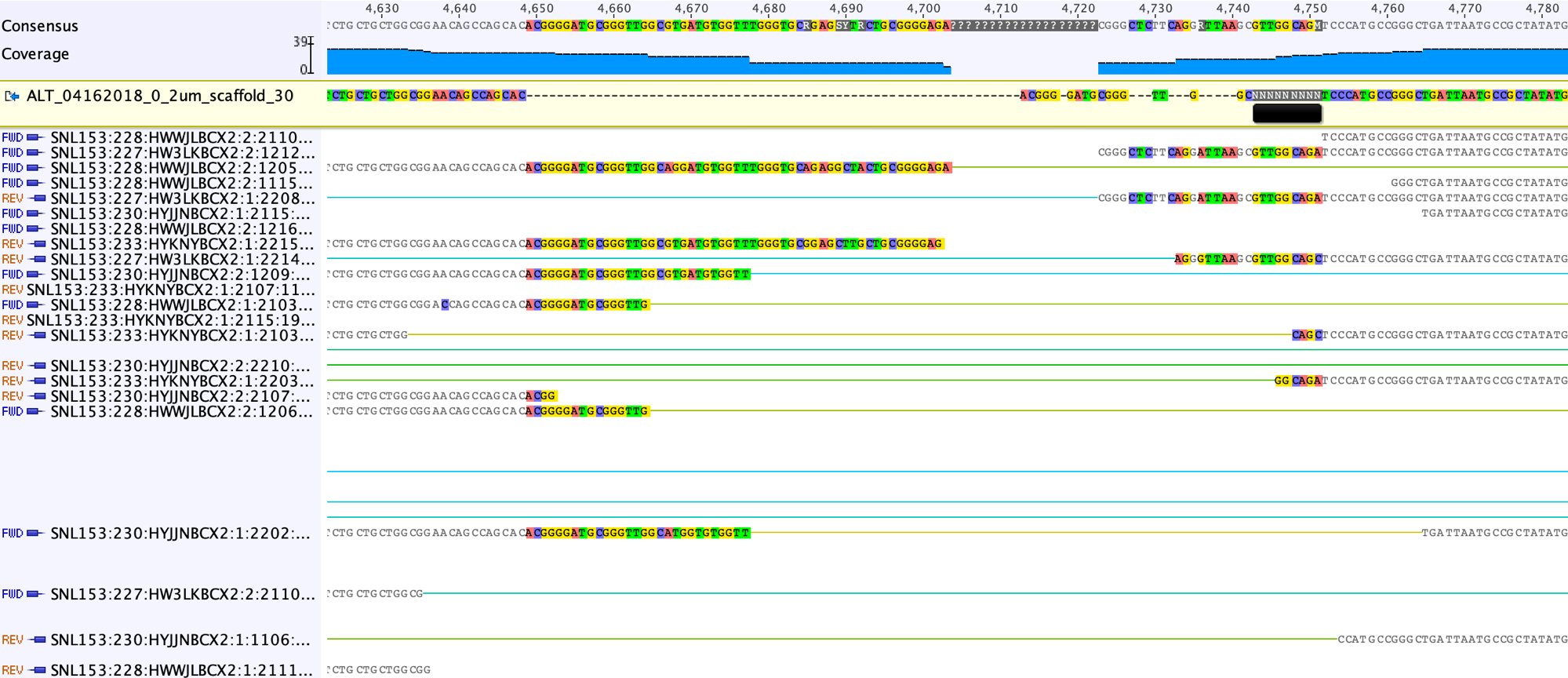
As you can see from the screenshot above, there are sequences shared by the reads that can be copied and pasted into the scaffold sequence to partially fill the gap.
Although the reads in the gap regions shared high sequence similarity, we can also see some single nucleotide variants (SNVs). We choose the consensus sequences shared by majority reads.
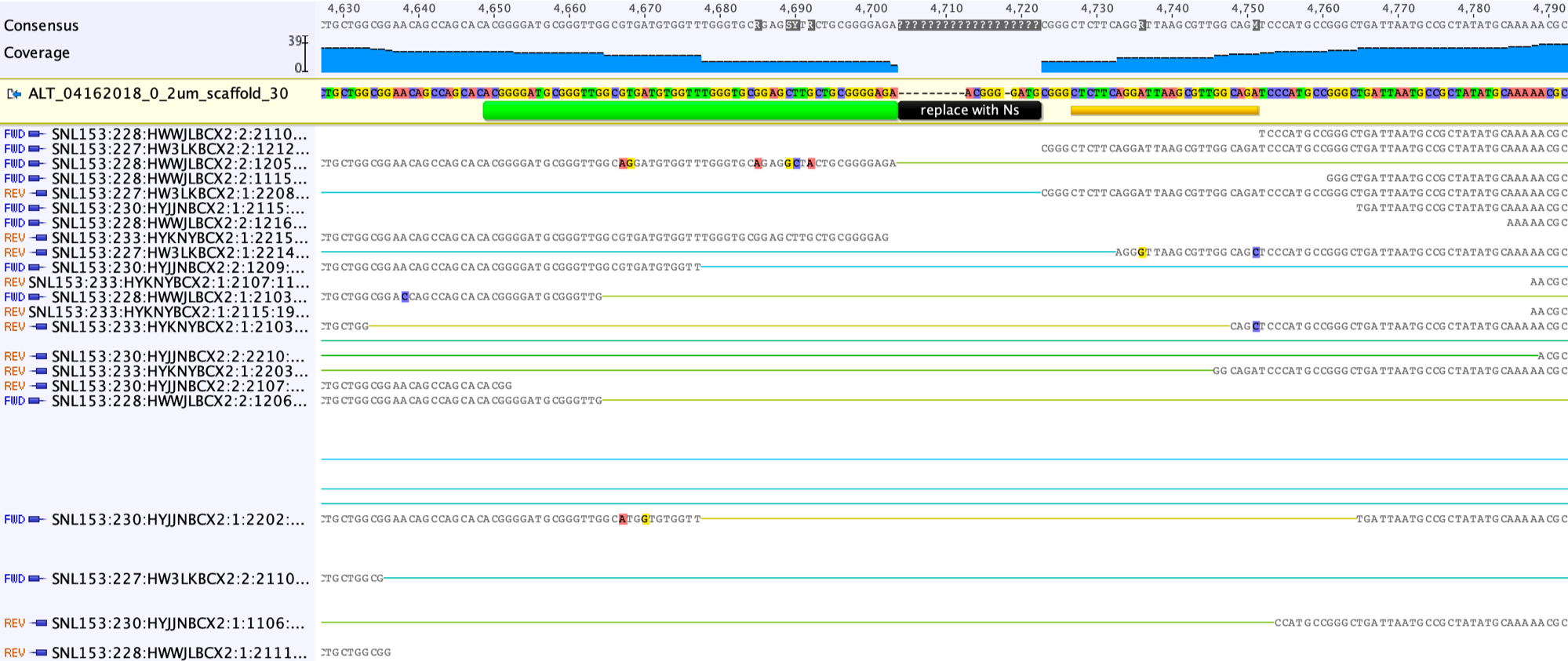
Unsupported consensus sequence should be replaced by Ns.
We then save the modified scaffold as “ALT_04162018_0_2um_scaffold_30_modified”. We map the unplaced paired reads (same as used to extend scaffold ends, see above) with the “Medium Sensitivity / Fast” to the modified scaffold.

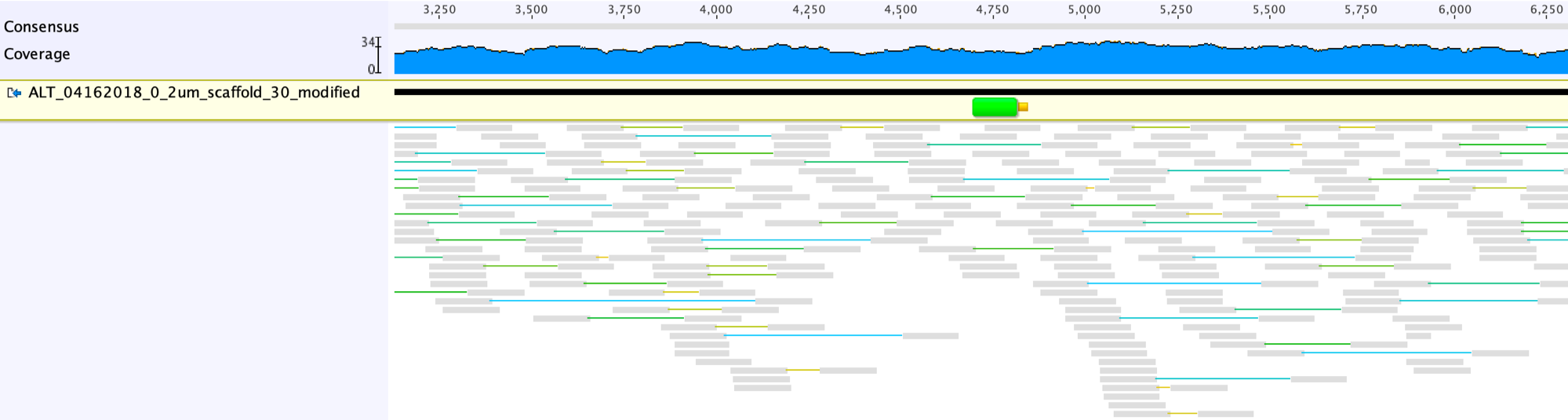
The output mapping profile of the region with the scaffolding gap is shown below:
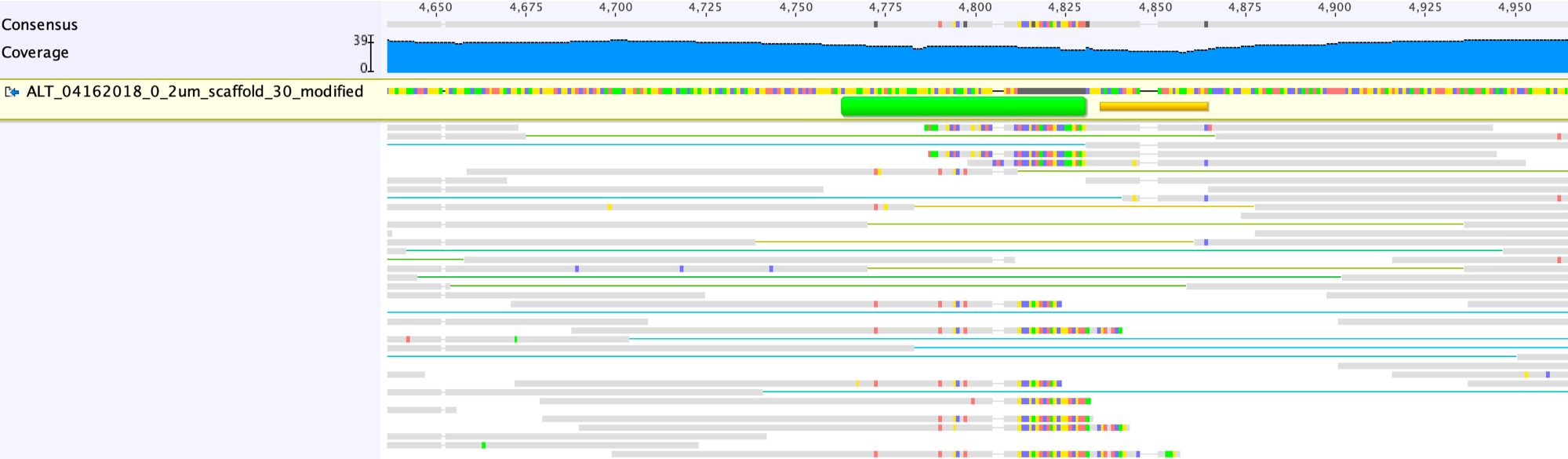
Open the gap and sort the reads as performed above.
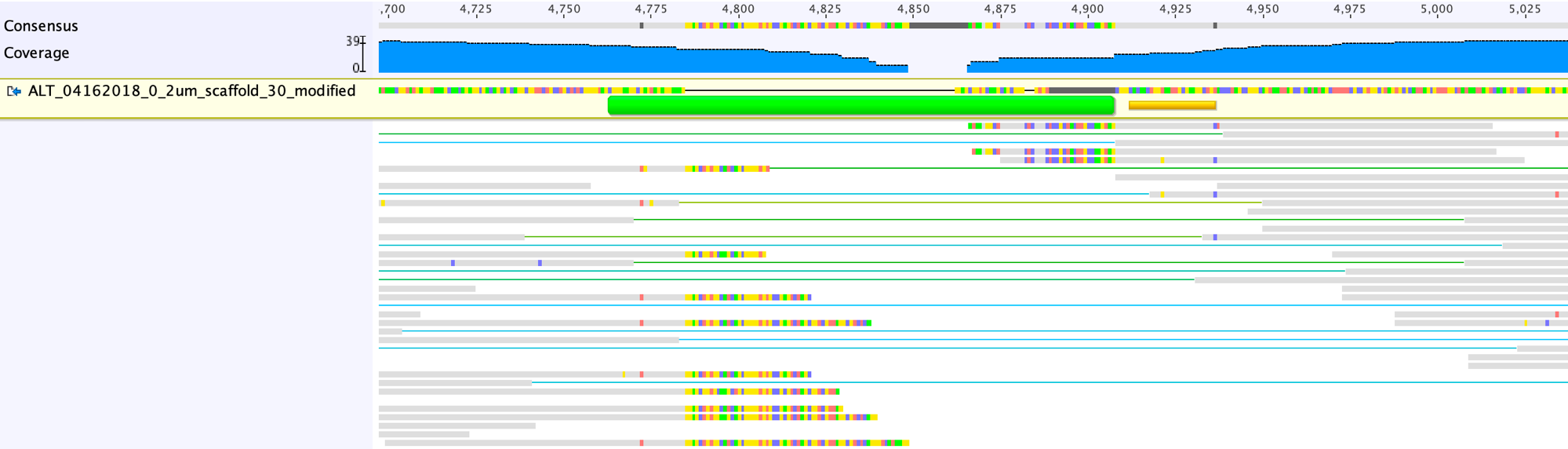
Add consensus sequences shared by the reads to the scaffold. Do not worry about the SNVs; we will figure out what is happening once the gap is closed.
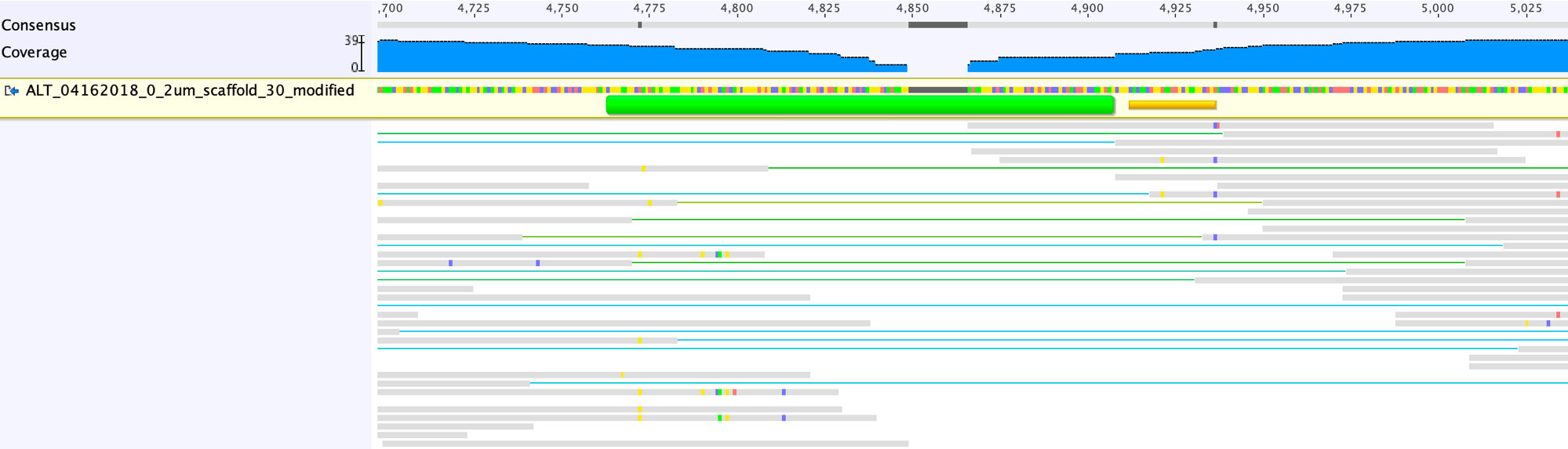
Run of mapping with unplaced paired reads until the gap can be closed:
The mapping profiles indicate the gap will be closed once we delete the bases in the scaffolds without read support:
Next, map the reads to the scaffold with the gap closed allowing no mismatch (custom sensitivity, zero mismatch allowed), to see if the region is covered throughout.
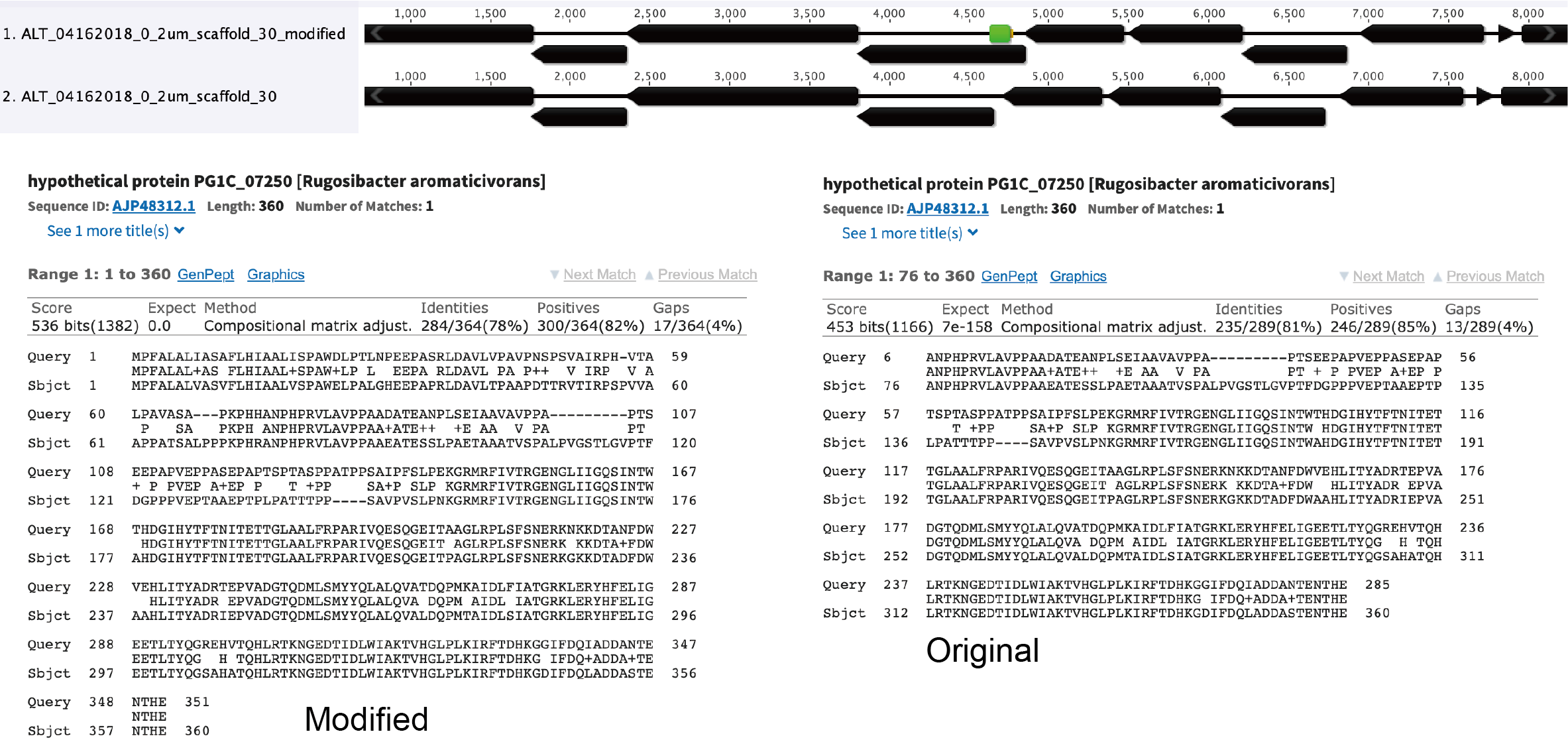
By closing the gap, we obtain a complete amino acid sequence for the relevant gene, which is confirmed by a BLASTp search:
Conclusions
In this blog, we show how the scaffold extension and scaffolding gap closing can be performed in Geneious. We hope that these insights could help users improve the quality of their MAGs. Importantly, automatic tools should be developed in the future to perform these curation steps. Please feel free to comment below or contact us via email if you are working on automated solutions or have different strategies or tips to improve MAGs.